Introduction
Xylella fastidiosa (Wells et al., 1987) is a pathogen of bacterial plants that cause many economically significant diseases, including grapevine disease of Pierce, citrus veinal chlorosis, almond leaf scorch, phony peach, and leaf scorch on a range of ornamental plants and shade trees (Hopkins and Purcell, 2002). X. fastidiosa is a globally regulated and banned pathogen. Leafhoppers of the subfamily Cicadellinae (Hemiptera: Cicadellidae) and spittle bugs or frog hoppers of the family Cercopidae (Hemiptera) are the most common known vectors (Purcell, 1997). The distribution of X. fastidiosa is usually restricted to the Americas (Purcell, 1997), with two exceptions, in Vitis vinifera in Kosovo (Berisha et al., 1998) and pear in Taiwan (Leu, 1993). X. fastidiosa is known to be vulnerable to low temperatures, which has confined its movement to regions with temperate climates and, in particular, to cold winters (Purcell, 1997). Many colder parts of the world, however, have one or more vector species, such as the spittlebug (Philaenus spumarius), so there is a possibility for X. fastidiosa to spread to such areas if strains are cold-tolerant, such as almond leaf scorch, become established (Purcell, 1997). From a quarantine viewpoint, the main aspect of any exclusion technique is rapid identification and diagnosis. Present X. fastidiosa diagnostic tests include bacterial cell culture, traditional polymerase chain reaction (PCR) (Huang, 2009; Huang et al., 2006; Minsavage et al., 1994; Pooler and Hartung, 1995; Rodrigues et al., 2003) and real-time PCR (Francis et al., 2006; Schaad et al., 2002). While many of these methods have been widely used in the laboratory, most of these methods are not readily transferable to the field. In addition, the PCR assay was developed over 15 years ago. When the X. fastidiosa DNA sequence was little usable, this assay is widely used for quarantine screening and, thus, checking that it reliably detects all isolates of the bacterium is especially essential. Alternative methods of detection have been considered in light of these factors. Loop-mediated isothermal amplification (LAMP) is one approach that has recently been implemented for plant pathogen diagnostics. It can be done in a heat block or water bath because the LAMP reaction is isothermal, thereby reducing the need for specialized equipment. Moreover, colorimetric or fluorescent dyes may be observed for positive amplification (Goto et al., 2009; Tomlinson and Boonham, 2008), removing the need to run gels. Both of these factors lead to the field’s transferability. The production and evaluation of a LAMP assay for X. fastidiosa is presented here in order to improve diagnostic capacity by allowing surveillance activities, improving response times during incursions, and enabling testing at the border for imported goods. During the development of the LAMP assay, the potential for developing PCR was based on the identification of the same region used for the LAMP primer design. A new PCR assay was also tested in accordance with the LAMP process.
Materials and Methods
Samples. X. fastidiosa DNA (ATCC 700964D-5, ATCC 35881D, ATCC 35881D-5) were obtained from commercial sources (American Type Culture Collection, Manassas, VA, USA). Control strains for specific diagnostic were obtained from commercial sources (Korea Agricultural Culture Collection, Suwon, Korea).
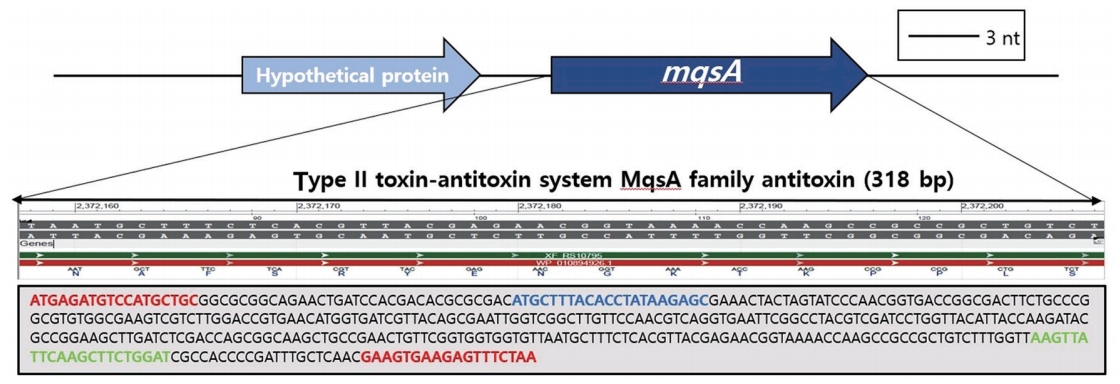
PCR primer design and PCR condition. PCR primers are the method to diagnose a large number of samples quickly, and fast and accurate diagnostics can be made without complicated equipment. After exploring different gene sites linked to the diagnosis of X. fastidiosa, primers were therefore prepared after the optimal target gene was selected (Table 1). The mqsA gene sequence of the standard strain X. fastidiosa was used to prepare PCR primers (Fig. 1). For PCR, the primer is 20 pM and AccuPower PCR premix (1 U Taq DNA polymerase, 250 μm dNTP, 10 mM Tris-HCI, 40 mM KCI, 1.5 mM MgCI2, stabilizer and tracking dye: Bioneer, Daejeon, Korea), template genomic DNA, a reaction solution with a total volume of 20 μl was prepared (Francis et al., 2006). Initial denature (2 min at 94°C), denature (1 min at 94°C), annealing (1 min at 55°C), extension (1 min 30 sec at 72°C) After reacting a total of 35 cycles, last extension (5 min at 72°C), reaction was carried out (Fukuta et al., 2003). Thereafter, the product was subjected to electrophoresis at 50 V for 40 min using 1.5% agarose gel added with SYBR Green I and 1× TAE buffer, stained with ethidium bromide, and observed under UV illumination.
LAMP primer design. In order to design the LAMP PCR primer, the primer was designed by referring to the 10 kinds of 16S rDNA nucleotide sequences registered in Gen-Bank. LAMP DNA oligonucleotide primer of X. fastidiosa is the BLAST program (Berisha et al., 1998). The following sequence was designed by selecting a primer for LAMP of X. fastidiosa using Primer Explorer version 3 (https://www.primerexplorer.jp/lamp3.0.0/index.html), a LAMP primer designing software (Fukuta et al. 2003). In the primer set, the following two inner primers (forward inner primer [FIP], backward inner primer [BIP]) that can recognize six specific regions of the target sequence composed of four primers are the complementary nucleotide sequence of the X. fastidiosa sequence and loop. It was produced by combining the partial TTTT spacer of the sequence forming the reverse transcription LAMP (Fukuta et al., 2003). The following two outer primers (F3: forward outer primer, B3: backward outer primer) were designed to be located outside the inner primer, respectively. A total of 6 primers were produced by ordering from Bioneer (Table 2).
Optimization of the XF 1535 gene LAMP assay. Twenty pM F3, B3 primer and 40 pM FIP, BIP primer, reagent 1× reaction mix (20 mM Tris-HCI [pH 8.8], 10 mM (NH4)2SO4, 10 mM KCI, 2 mM MgSO4, 0.4% Triton X-100, 6 mM MgSO4 [New England Biolabs, Beverly, MA, USA], 0.8 M betaine [Sigma-Aldrich, St. Louis, MO, USA], 400 μM dNTP [TaKaRa Biotechnology Co., Ltd., Tokyo, Japan]) was composed of a total of 25 μl of a reaction solution. The reaction solution was allowed to stand at 95°C for 5 min on the next heat block, and then cooled on ice, and 8 unit Bst DNA polymerase large fragment (New England Biolabs) was added. After that, the reaction solution was reacted at 65°C for 60 min and 80°C for 10 min (Fukuta et al., 2003) and visualized by adding SYBR Green I (Life Technologies, Chicago, IL, USA). This is orange in the absence of an amplicon, and green in the presence of the LAMP amplification product.
Sensitivity and specificity. Sensitivity and specificity were tested with Xanthomonas axonopodis pv. diffenbachiae, Pectobacterium atrosepticum, X. campestris, X. campestris pv. glycines, Pseudomonas syringae, Xaxonopodis pv. glycines, Burkholderia glumae, Burkholderia cepacia, Erwinia pyrifoliae, E. rhapontic, Pseudomonas tolaasii, Pseudomonas agarici, Pectobacterium carotovorum subsp. brasiliense, Pseudomonas cichorii, Burkholderia gladioli, Pseudomonas viridiflava, X. arboricola pv. prun, and X. campestris pv. vitians (Table 3). DNA (2.0×106 copies/ml) was diluted 10 times each, and the sensitivity and specificity of PCR and LAMP were tested at a concentration of 2.0×106 to 2.0×102 copies/ml.
Results
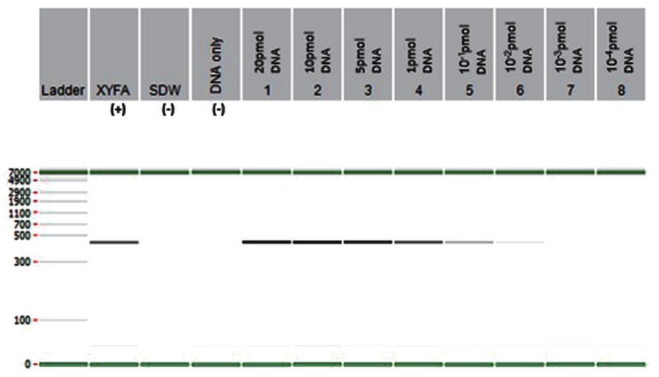
Specificity and sensitivity of created mqsA primer set. PCR was used to assess the specificity of the newly developed primer set. As a result of electrophoresis of the PCR product, the amplified result was obtained only in X. fastidiosa, and there was no non-specific reaction with other strains (Fig. 2). The mqsA site contained in toxin-antitoxin is a physiological regulatory site mainly involved in pathogenicity and motility, and it is considered to be highly useful as a species-specific primer for X. fastidiosa. Their detection ability was confirmed according to the PCR conditions (Fig. 3). The detection capability of Pierce’s disease bacteria X. fastidiosa was tested on 16 positive control and 16 negative control strains in total.
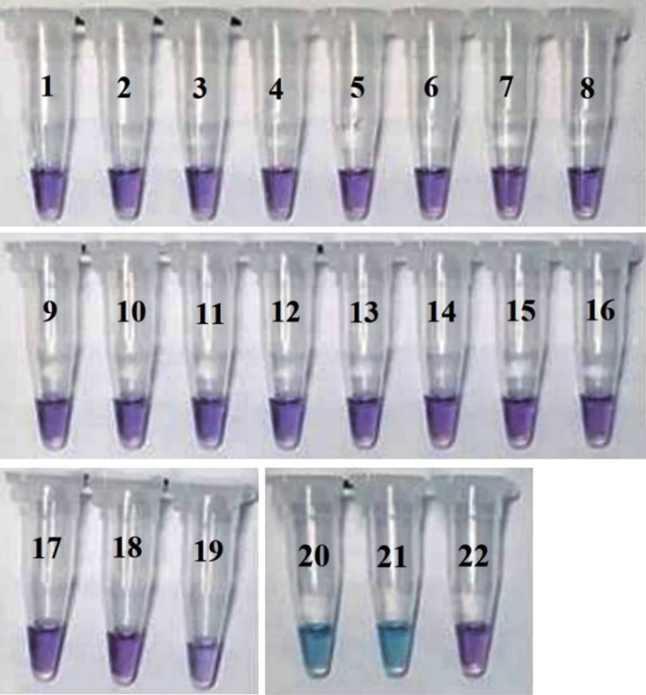
Confirmation of specificity of LAMP method and PCR method. As a result of testing the specificity of LAMP PCR using standard and similar strains, only X. fastidiosa showed a specific reaction (Fig. 4). In the case of LAMP PCR, it is necessary to observe the time because it may cause a false-positive reaction after the standard reaction time.
Confirmation of sensitivity of LAMP method and PCR method. After establishing the optimal conditions, the sensitivity of the LAMP detection method is determined by diluting the Streptococcus uberis culture solution to a concentration of 1.0×10 to 1.0×108 cfu/ml, and using the Genomic DNA Extraction Kit (Bioneer) After extraction, the detection limit values of the LAMP detection method and the PCR method were compared. In the case of PCR, it can be detected only up to a concentration of 1.0×10⁴ cfu/ml, while as a result of measuring the detection limit of LAMP, it was confirmed that it could detect up to a concentration of 1.0×10⁸ cfu/ml. It was confirmed that the LAMP method has a higher detection limit than the general PCR method.
In order to measure the sensitivity of the developed LAMP, the extracted DNA was diluted 10 times in steps, and then developed LAMP and PCR were performed on the genes extracted from each dilution. It was detected that the developed LAMP was 10 times more sensitive than the existing PCR (Fig. 5).
Discussion
LAMP PCR is a technology that uses basic laboratory equipment to diagnose a variety of research products, such as infectious diseases, in the field (Chen et al., 2008). This time the LAMP PCR method was developed for use in epidemic and quarantine research. The analysis shows that there was no difference from the conventional PCR method, because of its sensitivity and specificities (Fig. 3). Consequently, the findings of this study are considered sufficient for X. fastidiosa diagnoses with various hosts and symptoms. It is important to observe the time in the case of LAMP PCR, since it can cause a false positive reaction after the standard reaction time. For future more reliable and effective experiments, detailed analysis of the reaction conditions and reaction reagents is necessary.
In addition, microscopy, selective medium, and PCR techniques are primarily used in the diagnosis of plant diseases. Recently, by shortening the diagnosis time and using simple diagnosis methods away from competent and complicated diagnosis methods, there is a trend to create diagnostic kits. The LAMP PCR approach is not derived and amplified by the conjugation and expansion at isothermal temperatures and has the advantage, by using mainly 4-6 primers, of increasing the PCR specificity in the target species. Since DNA amplification is possible if the temperature is kept isothermal, only an isothermal maintenance system such as a water bath and heat block can be detected without an expensive PCR device. The detection result can be tested with the naked eye when a fluorescent dye is used, so it can be used immediately in the field, and the number of applications of the diagnostic method LAMP PCR is increasing in the field.
X. fastidiosa is a high-risk pathogen that causes disease in a wide variety of plants. Test methods have been developed in European and Mediterranean Plant Protection Organization and others to diagnose these pathogens (Notomi et al., 2000). However, in recent years, as new genetic knowledge on pathogens has been discovered, it is important to check existing test methods and to diagnose new methods. Therefore, a new test method was developed to complement the previously developed diagnostic method. The newly developed test method detects the mqsA gene of X. fastidiosa. The findings showed high specificity and sensitivity as a consequence of checking the specificity and sensitivity of 16 related strains (Figs. 2, 3). It is important to establish a quantitative test method (real-time PCR) for more accurate testing in the future.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print