Rice blast, caused by Magnaporthe oryzae, is the most devastating disease in rice worldwide (Ebbole, 2007) and destroys 10-30% of rice production annually (Talbot, 2003). As over half of the global human population depends on rice as staple food (Strange and Scott, 2005), intensive research has been conducted to understand its mechanism of pathogenicity towards the goal of developing effective control strategies for the rice blast.
The infection of rice by M. oryzae is initiated by conidial attachment on host surfaces followed by conidial germination. Via recognition of physical cues on the rice surface, such as hydrophobicity and surface hardness, emerging germ tubes initiate formation of a specialized infection structure, called the appressorium, within 4-6 hours (Hamer and Talbot, 1998; Howard and Valent, 1996). The appressorium is a dome-shaped cell that enables the fungus to penetrate the rice leaf surface. Enormous turgor pressure (8 MPa) builds up inside the melanized appressorium due to high glycerol concentration, which drives penetration of the plant surface. After successful infection of plant tissues, visible symptoms appear rapidly. Over two decades of research has unequivocally established the involvement of several signaling pathways, including the cyclic AMP-dependent (Adachi and Hamer, 1998; Lee and Dean, 1993; Mitchell and Dean, 1995; Xu and Hamer, 1996), calcium-dependent (Choi et al., 2009a, 2009b; Liu and Kolattukudy, 1999; Nguyen et al., 2008), and mitogen-activated protein (MAP) kinase signaling pathways (Hamer and Talbot, 1998; Jeon et al., 2008; Soanes et al., 2012; Thines et al., 2000; Xu and Hamer, 1996) in appressorium differentiation.
One notable advantage that has facilitated studies on appressorium differentiation is the possibility to induce appressorium formation on artificial hydrophobic surface as well as hydrophilic surfaces in the presence of cAMP (Lee and Dean, 1993). Multiple approaches have been deployed to identify genes important in M. oryzae appressorium development, including suppression subtractive hybridization (Lu et al., 2005), super serial analysis of gene expression (Irie et al., 2003; Soanes et al., 2012), expressed sequence tag analysis (Oh et al., 2008), and RNA-sequencing (Soanes et al., 2012). Identification of genes expressed in appressoria via such approaches and subsequent functional analysis of identified genes via mutagenesis have helped understand the molecular mechanism underpinning the initiation and maturation of M. oryzae appressorium. Even though a large number of genes involved in appressorium differentiation have been identified via functional validation, many more genes likely remain to be identified.
In this study, we applied cDNA-amplified fragment length polymorphisms (cDNA-AFLP) analysis (Park et al., 2009) to identify and characterize the function of selected genes using the previously constructed ATMT mutant library (Jeon et al., 2007).
M. oryzae strain KJ201 (wild-type) was obtained from the Centre for Fungal Genetic Resources at Seoul National University, Seoul, Korea. All fungal isolates were grown on V8-juice agar (8% V8 juice, pH 6.8) or oatmeal agar (5% oatmeal [w/v], 2% agar [w/v]) at 25°C under constant light to promote conidial production. The wild-type and ATMT mutants were cultured on complete agar medium, minimal agar medium, C starvation medium, and N starvation medium to observe mycelial growth and colony characteristics (Talbot et al., 1997).
Total RNA was isolated from appressoria, formed on hydrophobic glass for 4-6 hr, and mycelia cultured in complete liquid medium. Extracted RNA (5 μg) was used to produce random primed cDNA with the Prom-II Reverse Transcription System (Promega, Madison, WI, USA). Three independent experiments were performed.
This analysis was performed as previously described (Park et al., 2009). cDNA (200 ng) was digested with EcoRI and MspI and ligated to EcoRI and MspI double strand adapters (Park et al., 2009). The products were diluted (50×) with TE buffer and 5 μl were used for selective amplification, using 36 cycles that include 13 touchdown cycles comprising a reduction of the annealing temperature from 65°C to 56°C in 0.7°C steps, which were subsequently maintained for 23 cycles. A total of 28 primer combinations (Table 1) were used for selective amplification as previously described (Park et al., 2009).
Table 1
Primers used for cDNA-AFLP
Bands of interest were selected, excised from the gel, and re-amplified as previously described (Park et al., 2009). PCR products were confirmed on 1.0% agarose gels. The amplified transcript-derived fragments (TDFs) were cloned into the plasmid pGEM-T Easy vector system I (Promega). Sequencing of cloned TDFs was conducted on an Applied Biosystems sequencer (ABI3700, Applied Biosystems, Foster City, CA, USA).
Database searches were performed using the BLAST Network Service (NCBI, National Center for Biotechnology Information) and the Magnaporthe genome database (http://www.broad.mit.edu/annotation/fungi/magnaporthe/). To determine whether the genes derived from TDFs were orphan genes or had orthologs in other organisms, the BLAST Matrix program incorporated in CFGP (Park et al., 2008) and InParanoid algorithms (Remm et al., 2001) were used. We applied the cutoff e-value of less than 10-50 for protein similarity in BLAST Matrix searches and the InParanoid default parameters.
Quantitative real-time RT-PCR (qRT-PCR) was applied to measure transcript levels using specific primers (Table 2) and the conditions described previously (Park et al., 2013). All reactions were performed with three biological and three technical replicates for each sample. The β-tubulin gene was used as the internal control for normalization. All amplification curves were analyzed with a normalized reporter threshold of 0.1 to obtain the threshold cycle (Ct) values. The comparative ΔΔ Ct method was applied to evaluate relative quantities of each amplified sample product. Fold changes were calculated as 2−ΔΔCt (Livak and Schmittgen, 2001). We applied a fold-change cutoff of ≥1.5 for up-regulation and ≤0.5 for down-regulation.
Table 2
Primers used for quantitative real-time PCR
Isolation of differentially expressed TDFs
To isolate TDFs corresponding to differentially expressed genes during appressorium development in M. oryzae, cDNA-AFLP analysis was performed using RNAs extracted from appressoriumforming conidia on hydrophobic glass and vegetatively grown mycelia in complete liquid medium.
A total of 28 primer combinations were used (Table 1). Resulting fragments were ranged in size from 50 to 1,200 bp. Over 200 TDFs that appeared to be differentially expressed genes were detected. Each band was directly excised from the gel, eluted, and re-amplified using the same primer set. Among the 200 TDFs, 52 were successfully cloned and sequenced. In order to obtain more up-regulated or differentially expressed genes during appressorium development, additional primer combinations for cDNA-AFLP could be helpful.
Identification of up-regulated genes during appressorium formation
The GenBank nucleotide sequence database (http://blast.ncbi.nlm.nih.gov/Blast) and Magnaporthe database (http://www.broad.mit.edu/annotation/fungi/magnaporthe/) were queried using sequences of the 52 TDFs via the BLAST (Basic Local Alignment Search Tool) program. The majority (44 TDFs) matched to M. oryzae genes (Table 3). Based on blast search, we identified 42 independent genes that are likely associated with appressorium development. Among 44 TDFs, 2 TDFs are identical sequence to two TDFs which identified as MGG_01117T0 and MGG_15150T0, respectively.
Table 3
List of 44 transcript-derived fragments corresponding to 42 genes and the relative abundance of their transcripts during appressorium development
The genes up-regulated during appressorium development included the following: homologues of the dynactin Arp1 P62 subunit RO2 (MGG_00222T0) (Lee et al., 2001), which regulates the activity of cytoplasmic dynein (a microtubule-associated motor); a gene encoding mitogen activated protein kinase kinase kinase3, MCK1 (MGG_00883T0) (Jeon et al., 2008), which is an integral component of the MAP kinase signaling pathway that regulates cell wall integrity in Saccharomyces cerevisiae (Levin, 2005) and M. oryzae (Jeon et al., 2008); a gene homologous to S. cerevisiae Git1 (MGG_00464T0), which encodes a permease involved in glycerophosphoinositol uptake (Patton-Vogt and Henry, 1998); beta-hexosaminidase (MGG_00621T0) homologue; a homologue of glycosylphosphatidylinositol-anchored extracellular cell wall glucanase Crf1 of Aspergillus fumigatus (MGG_10431T0) (Arroyo et al., 2007); and a pp-loop family protein (MGG_17376T0) homologue (Table 3).
The genes specifically expressed during appressorium development also included a gene putatively encoding acetylglutamate synthase (MGG_01507T0), a gene involved in autophagy; ATG6 (MGG_03694T0), which encodes a protein that forms a membrane-associated complex with ATG14p in S. cerevisiae and is critical for M. oryzae pathogenicity; protein phosphatase 1K (MGG_03918T0); L-ornithine 5-monooxygenase OMO1 (MGG_04212T0); cation efflux family protein (MGG_04407T0); potassiumactivated aldehyde dehydrogenase (MGG_05814T0); nuclear cap-binding protein subunit2 (MGG_06296T0); transport protein SEC23 (MGG_06910T0); CCR4-NOT transcription complex (MGG_08101T0); XPG N-terminal domain-containing protein (MGG_08303T0); cystathionine gamma-lyase (MGG_10380T0); ceramide glucosyltransferase (MGG_10668T0); and DUF124 domain protein (MGG_11494) (Table 3).
Among the 42 genes, only seven (MGG_00222T0, MGG_00464T0, MGG_03694T0, MGG_04538T0, MGG_10380T0, MGG_11809T0, and MGG_15282T0) had already been identified in a previous study (Soanes et al., 2012). The remaining 35 genes were not previously captured in appressorial transcriptome studies in M. oryzae (Oh et al., 2008; Soanes et al., 2012). This low redundancy level compare to other previous transcriptome analysis (Oh et al., 2008; Soanes et al., 2012) might be due to one or more of the following differences among these experiments: (1) differences in experimental methods; (2) inductive surface and treatments for appressorium development; (3) time points for sample collection; (4) differences between strains used; and (5) Magnaporthe genome versions used.
To determine whether the 42 genes have orthologs in other species, we applied BLAST Matrix (http://cfgp.snu.ac.kr) and InParanoid (Remm et al., 2001) to detect orthologs (Fig. 1). We found 5 genes (MGG_14593T0, MGG_15150 T0, MGG_14683 T0, MGG_14684 T0, and MGG_14741 T0) were M. oryzae-specific, based on the absence of orthologs in other species (BLAST E < 10-50 and InParanoid) (Fig. 1). Further examination is needed to determine whether these genes play an important role during M. oryzae appressorium devel opment and/or pathogenesis.
Fig. 1
Distribution of 42 appressorium-specific genes analyzed using BLAST Matrix and InParanoid algorism. Abbreviation for fungal species and other organisms: Pi, Phytophthora infestans; Mgr, Mycosphaerella graminicola; Af1, Aspergillus fumigatus Af293; An, Aspergillus nidulans; Ci, Coccidioides immitis; Hc8, Histoplasma capsulatum H88; Bg, Blumeria graminis f. sp. hordei; Bc, Botrytis cinerea; Cg, Colletotrichum graminicola M1.001; Fg, Fusarium graminearum; Fol, Fusarium oxysporum; Mo, Magnaporthe oryzae 70-15; Nc, Neurospora crassa; Pa, Podospora anserina; Cas, Candida albicans SC5314; Sc, Saccharomyces cerevisiae S288C; Sp, Schizosaccharomyces pombe; Hi, Heterobasidion irregulare TC32-1; Lb, Laccaria bicolor; Pc, Phanerochaete chrysosporium; Sl7, Serpula lacrymans S7.3; Ecu, Encephalitozoon cuniculi JAM81; Pbl, Phycomyces blakesleeanus; Ro, Rhizopus oryzae; Dm, Dorosophila melanogaster; Hs, Homo sapiens; Ce, Caenorhabditis elegans; Ath, Arabidopsis thaliana; Osj, Oryza sativa japonica. Red bar, M. oryzae-specific; orange bar, Ascomycota-specific; blue bar, fungal-specific; gray bar, conserved in whole organisms that we used in this study.
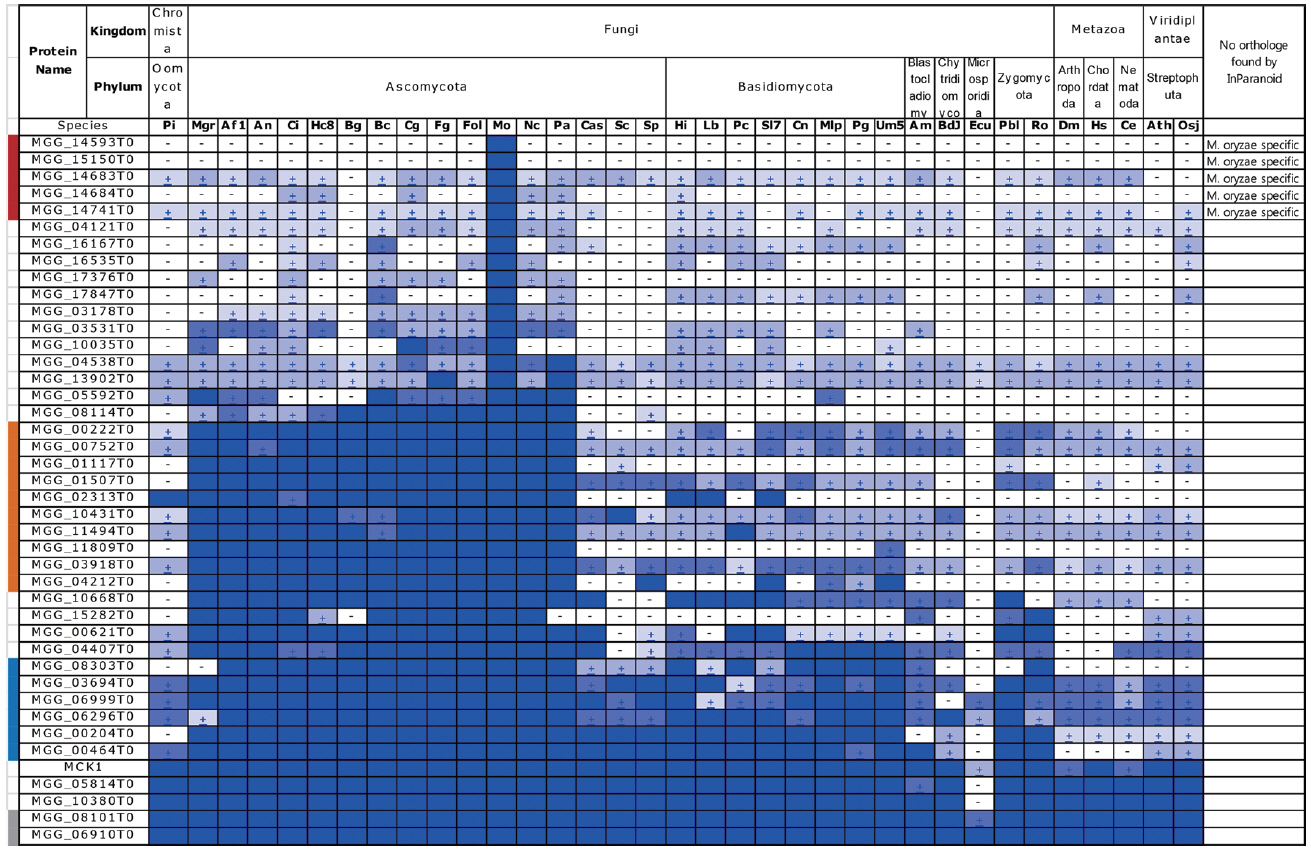
We also found that 10 genes (MGG_00222T0, MGG_00752T0, MGG_01117T0, MGG_01507T0, MGG_02313T0, MGG_10431T0, MGG_11494T0, MGG_11809T0, MGG_03918T0, and MGG_04212T0) were Ascomycota-specific (particularly in Pezizomycotina), 6 additional genes (MGG_08303T0, MGG_03694T0, MGG_06999T0, MGG_06296T0, MGG_00204T0, and MGG_00464T0) were fungal-specific, and 2 genes (MGG_08101T0 and MGG_06910T0) were conserved in all organisms included in the analysis (Fig. 1).
Validation of expression patterns by qRT-PCR
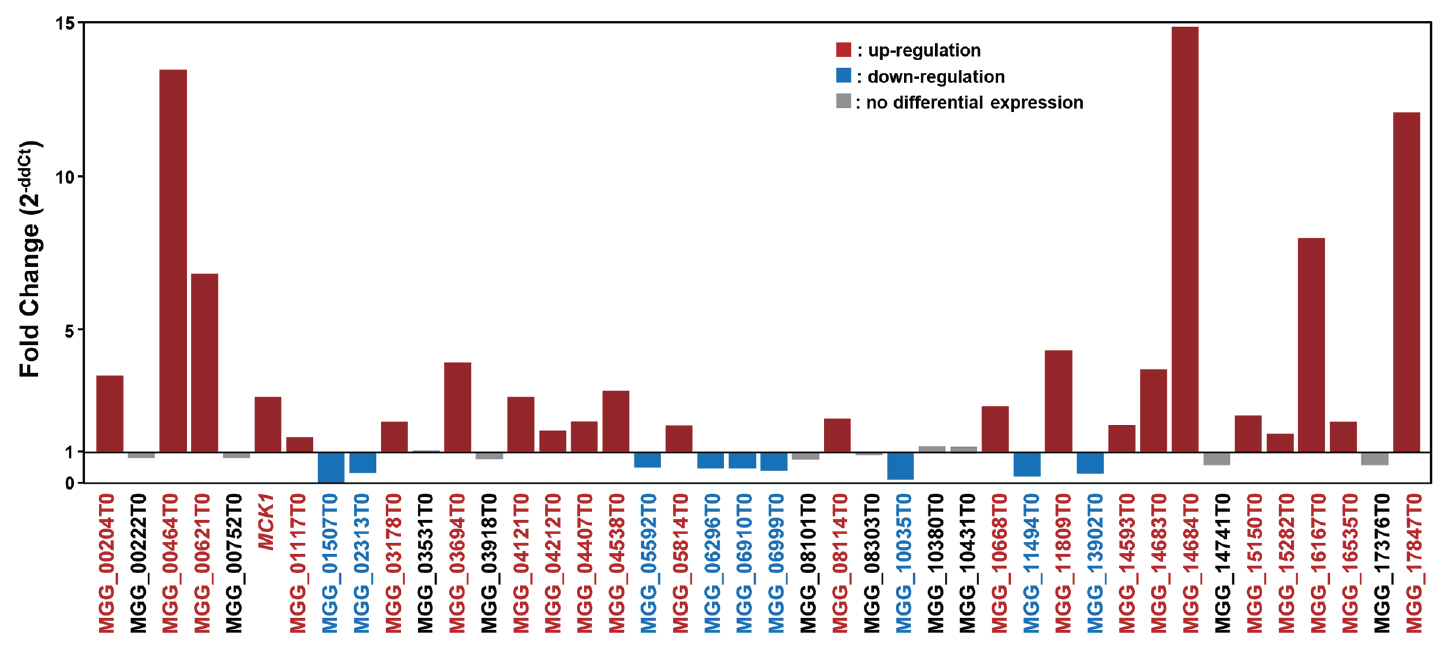
To validate gene expression patterns suggested by cDNA-AFLP analysis, qRT-PCR analysis was conducted. A total of 23 genes (54.8%) were up-regulated, 9 genes (21.4%) were down-regulated, and 10 genes (23.8%) were not differentially expressed (Fig. 2).
Fig. 2
Quantitative real-time PCR analysis of gene expression during appressorium development. Up-regulated genes (more than 2 fold) are indicated by red bars, down-regulated genes (less than 0.5 fold) are marked by blue bars. The genes did not show differentially expression are noted in black.
MGG_04212T0 encodes an L-ornithine 5-monooxygenase (OMO1), which is the key precursor for all hydroxamate siderophore biosynthesis in fungi (Eisendle et al., 2003; Schrettl et al., 2007). The siderophore is known to be required for virulence in A. fumigatus (Schrettl et al., 2007). We found that OMO1 expression was significantly up-regulated during appressorium formation (Fig. 1). Previous studies revealed that overcoming host-driven oxidative stress was a prerequisite for full pathogenicity in rice blast fungus (Chi et al., 2009; Huang et al., 2011). We previously showed homeostatic regulation of ferrous ion might serve an important role in pathogenicity via overcoming oxidative stress (Chi et al., 2009).
Similarly, MGG_04407T0, which encodes a putative cation efflux protein, and is also required for regulation of metal ion concentration. Further investigation is required to elucidate the roles of MGG_04212T0 and MGG_04407T0 in M. oryzae pathogenicity and metal ion regulation.
cDNA-AFLP, sequencing and qRT-PCR can be combined for effective identification of differentially expressed genes under specific conditions. Using these approaches, we have identified and confirmed differentially expressed genes during appressorium formation in M. oryzae. Subsequent expression, bioinformatics, and functional validation revealed interesting pathogenicity-related genes in M. oryzae. Further gene knock-out analysis would determine the role of these genes in appressorium development and/or pathogenicity.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print